
What Death? The Glaringly Obvious Secondary Endpoint Error on the COMIRNATY Label
Proof that the FDA Rubber Stamped the Pfizer BLA with Little to No Scrutiny
On August 23rd the FDA released the label for the new BLA granted to Pfizer for its COMIRNATY vaccine that states on page 18 in Table 6 there was only 1 “First Severe COVID-19 Occurrence” based on the FDA’s predefined criteria for evaluating this secondary endpoint. In the same table it says that there were 0 according to the CDC criteria.
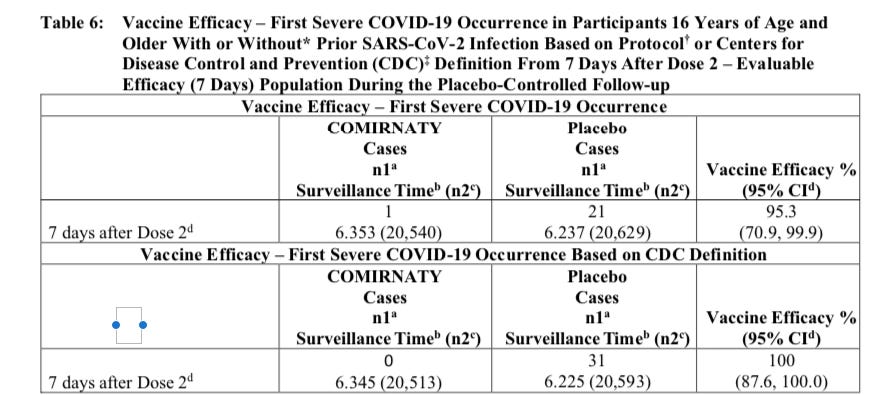
Source: COMIRNATY Package Insert, page 18, Table 6
Note that Pfizer is presenting data on patients 7 days after dose 2 in the above graph. Just to make my point absolutely clear, I want to point out that Pfizer is still essentially reporting that same 1 case in everyone who got the drug after dose 1 as well in the recent presentation of a pre-print of their 6 month follow up:
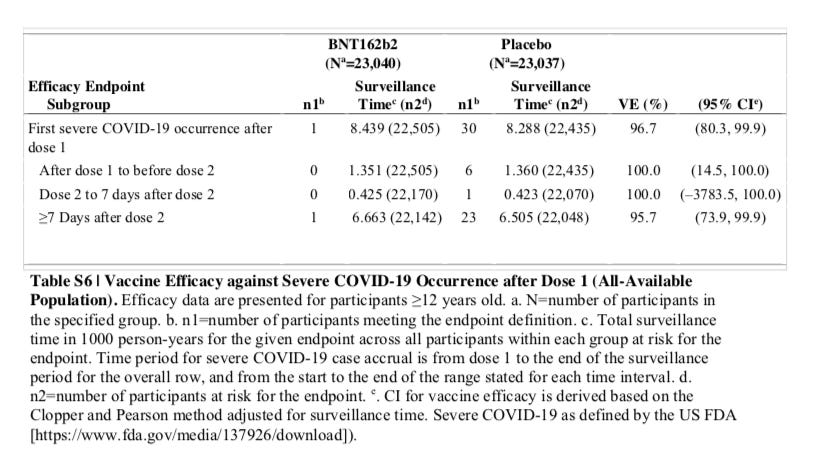
Source: SUPPLEMENTARY APPENDIX to Six Month Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine, Page 15, Table S6
This table from the supplementary appendix corresponds to the first column of the table in the package insert which is based on the pre-defined FDA protocol, not the CDC definition. If you move the timeframe back to the first dose, Pfizer still is reporting only one severe COVID-19 case. Basically there are only 7 cases that were identified in the placebo group prior to the week after dose 2, and according to Pfizer none were in the vaccine group. Note also that this table contains more patients and events since it includes ages 12 and up, while the label only addresses the formal BLA approved ages of 16 and up.
Also note that all patients were included whether they had evidence of prior exposure to COVID-19 or not in the first table, which is not consistent with the originally defined secondary endpoint which was supposed to focus on those without exposure to COVID-19 as can be seen in the analysis as it was supposed to appear in the panel briefing document on the earlier, less complete, in Table 11:

Source: VRBPAC 12/10/2020 Briefing Document, page 31, Tables 11 and 12
You can see in Table 11 that it says “without evidence of prior SARS-CoV-2 infection” on the original secondary analysis, just like the primary analysis. Table 12 is where they add in patients with with evidence of prior SARS-CoV-2 infection to get 1 versus 4 instead of 1 versus 3. This is a side point, I just wanted you to notice how the FDA let them change how they displayed their secondary efficacy endpoint to very slightly boost the results in their favor in the final BLA. That was utterly unnecessary, unusual, and if they were going to do that, why not at least do it with more data as it was presented on page 31 in the original briefing document.
The CDC criteria was not even put in the original briefing document, as you can see, because that is not what the FDA told them to look for. The sponsor did bring this data up at the FDA Panel Meeting because they were desperate to make the data look better because they missed their original secondary endpoint at the first analysis presented to the panel. I have to admit, I like that criteria better as well as a hospitalization is a better indication of severe disease than a pulse oximeter reading of below 93 and a high heart rate. The FDA probably had a problem with a hospitalization being subjective, and I can see that point of view as well. An ICU admission is a lot less ambiguous - that is not just severe, but a critical COVID-19 case.
That is not my main point. I am just trying to demonstrate here more evidence of how the FDA was bending over backwards to accommodate Pfizer. I rarely see this, but it has been a while admittedly since I listened to my last panel meeting.
I just want to establish if you look at all the data, both on the label and in the 6-month follow up that they are claiming according to the FDA criteria, only 1 person had severe COVID-19, and that was both at dose 1, and 7 days after dose 2, with and without evidence of prior infection. It is not like there is an event that is missing because you went from dose 1 to 7 days after dose 2 as an out for what I am about to show you.
The problem is, based on other published reports of the data if they are also to be believed, this cannot possibly be true. The number severe COVID-19 cases in the secondary endpoint has to be at least 2, under the FDA criteria, not 1.
We know from the document submitted to the FDA Advisory panel in December that there was one “first severe COVID-19 occurrence” in the vaccine arm as can be seen on page 31, Table 11 of that document (shown above). Of this particular case, it was said right above this table on the prior page, “The vaccine recipient who had severe COVID-19 disease met the severe case definition because oxygen saturation at the COVID-19 illness visit was 93% on room air. The subject was not hospitalized, did not seek further medical care, and did not have risk factors for severe disease.”
You can see it for yourself here:

Source: VRBPAC 12/10/2020 Briefing Document, page 30, Severe COVID-19 Cases.
So this patient clearly did not die, and would be consistent with the one patient shown on the FDA label that was not classified as severe COVID-19 as per the CDC standards.
A death on the other hand, would be considered “severe” by both criteria. And just in case there is any doubt on that, here is the footnotes to the tables I showed you above that explain both the predefined FDA criteria for severe COVID-19 and the CDC criteria:

Source: COMIRNATY Package Insert, page 18, Footnotes to Table 6
Death is listed clearly under both criteria as the last bullet point under each heading. So if you have a COVID-19 patient, that dies as a result of the condition, that person has to be counted as a severe COVID-19 case. I would not think anyone would have to even say that. A death is about as severe as it gets. But, apparently not for Pfizer investigators…
And a COVID-19 death is exactly what we have in the vaccine group when you look at the pre-print for the 6 month followup data from Pfizer. On the supplementary tables on page 12, Table S4, “Reported Cause of Death” in the vaccine arm there is exactly one “COVID-19 pneumonia” listed as cause of death. Interestingly enough, there are two cases of “COVID-19” listed as a cause of death for the placebo arm.
Look for yourself in the table below:
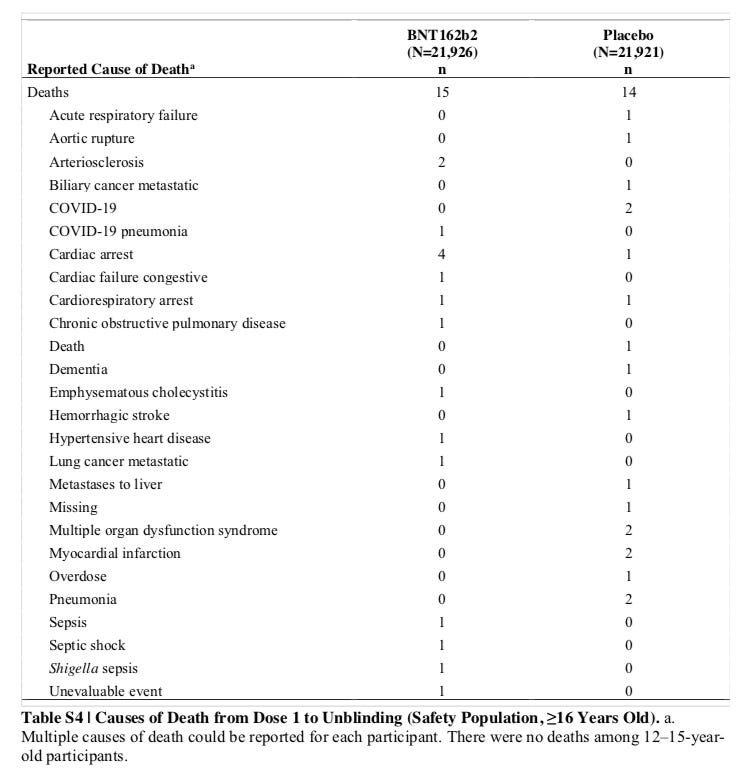
Source: SUPPLEMENTARY APPENDIX to Six Month Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine, Page 12, Table S4
Please note that Table S4 above says to “to Unblinding” and the Table 6 on the label says “During the Placebo-Controlled Follow-up.” I am trying to figure out any artful use of words that Pfizer might try to come up with and explain this away. But to most people “unblinding” is the exact end date of the “placebo-controlled follow-up.” The best case the FDA has in my opinion is to claim the death happened from dose 1 to 7 days after dose 2 and it was Pfizer that lied in its six-month report, and not the FDA. That is an explanation I would not be buying though given (1) Pfizer itself lied showing the data itself was not sound to begin with and (2) in December they had 21,314 evaluable patients in the first report (see Table 12) with that same 1 adverse event and there were no COVID-19 deaths reported in either arm at the time. So most of their total 21,926 patients in the vaccine arm (COMIRNATY Package Insert, page 12), were already past the second dose without a death, and a big part of the difference between Tables 12 and 11 would have been the people Table 11 excluded not for making the one week after dose 2 benchmark, but for showing prior COVID-19 exposure. I am just not buying it.
The most likley scenario is Pfizer took the inaccurate severe COVID-19 data from dose 1 then presented in the preprint and the FDA just rubber stamped it.
A COVID-19 pneumonia death is a COVID-19 death regardless of what is on the certificate. If I wanted to be cynical I might say that Pfizer intentionally had this listed as a “COVID-19 pneumonia” death instead of a “COVID-19” death because they somehow think that way it would not count as a “COVID-19 death” and thus not need to be included in the severe COVID-19 cases. However, you would not expect the FDA or any independent arbiter to let them get away with that. If a person dies as a result of COVID-19, that is a COVID-19 death. End of story, you would think.
However, ignore this very “COVID-19 pneumonia” death is exactly what they did! The pulse ox case and the “COVID-19 pneumonia death” equals 2 severe COVID-19 cases total, not one as per FDA criteria and one for CDC criteria, not the zero reported.
Severe COVID-19 was a pre-defined secondary endpoint of the trial; see page 30 of the FDA panel briefing document: “The secondary efficacy endpoints evaluate the VE of BNT162b2 for the prevention of COVID-19 disease from 14 days after Dose 2 and based on the CDC’s definition of COVID-19 disease from 7 and 14 days after Dose 2.” The secondary and primary endpoints literally should get the most scrutiny of all the data submitted since those are the most important pre-specified endpoints. Teams of people within the FDA are supposed to be given the job of scrubbing through this data to make sure it is accurate.
It is just unbelievable to me that given any normal review, a mistake that is as OBVIOUS as this one could be made if an ACTUAL REVIEW were undertaken by the FDA to make sure Pfizer was not fudging the data.
And this is based on the data we actually have! How many mistakes are there if we got to see the full data set the FDA presumably has access to?
How many of the 6,947 events in the vaccine arm (4,396 in ages 16-55 plus 2,551 in 56 and older, see page 12 of the label) should have been labeled “severe” but were not because Pfizer was trying to minimize the severity of the effects?
How many of the 262 severe adverse events listed in the pre-print (page 11, table S3) should have been labeled serious, because Pfizer did not consider them appropriately disabling to normal lives? Why do we not see number of severe adverse events on the label at all?!
Did Pfizer even use the normally accepted definitions for “severe” vs. “serious” you would expect in an FDA study? I would love to be on a panel and ask them specific questions about how they would grade certain adverse events like this one and many of the other horror stories that social media is looking to scrub from the internet. I would not even be totally shocked if they did not even classify something like this as “severe” given a “death” is not even “severe” in the minds of the Pfizer investigators.
If they underreported at least one severe COVID-19 cases, are there other ways that Pfizer avoiding reporting to boost the difference between treatment and placebo arms?
In the first report to the FDA panel, there two “suspected COVID-19” hospitalizations, both in the vaccinated group that tested RT-PCR negative, which is exactly what you might expect if a virus mutates, as the PCR primers test were designed against the RNA genetic code of very specific strains of COVID-19.

Source: VRBPAC 12/10/2020 Briefing Document, page 42, Suspected COVID-19 Cases
These two potential COVID-19 cases appeared as adverse events instead of severe COVID-19 as per the CDC criteria (it is not clear if they would meet the protocol criteria), and to be fair, one probably is just a reaction to the vaccine; but that is almost a worse outcome from the manufacturer’s perspective.
I am quite curious to know if there were other severe suspected COVID-19 cases or hospitalizations that were not listed due to a RT-PCR negative test at 6 months like there were on the interim report given to VRBPAC in December. That information is not on the label, and it would be quite interesting to know in terms of real world efficacy. I have another article I plan to write on that very subject, and it is something I am shocked no one else has looked at since the original briefing document came out in December 2020 because this was one of the first things that stuck out in my mind.
The Credibility of the FDA Irreparably Tarnished
The U.S. Food and Drug Administration (FDA) is expected to be an independent mediator of data generated by manufacturers in registration studies. After all, every drug company has a financial incentive to doctor the numbers in order to gain approval of a medicine.
But this does not always happen. Sometimes agency decisions are driven by political considerations given drug companies give lots of money to politicians of both parties.
Don’t believe me. Listen to the testimony of Dr. David J. Graham who spoke before Congress on this issue in 2004 after spending 20 years with the FDA talking about drugs that should have never been approved.
This was not a shock to me, because I was told the same thing by a consultant who focused on FDA issues in 2002. I was told that George W. Bush was furious Imclone’s first BLA was turned down despite a poorly done trial, and that his administration exerted pressure on the agency to approve other drugs after this.
I had also seen drugs that should not have been approved prior to that. I told my parents not to take COX-2s in November 2001 right after seeing the original medical review document on Bextra; the medical reviewer expressed concerns with the 7-fold increase in thromboembolic events despite naproxen given in both arms (which Merck falsely speculated was cardioprotective in their trials) of a high dose sub-study. It should have never been approved, and it was only 3 years later both Bextra and Vioxx were taken off the market.
Aside from corporate profits for large political donors, having a pharmaceutical approved might even align with certain government goals, such as more control over people using a Nazi-like scheme of vaccine passports to control the movement and ability of people to conduct commerce.
It is like someone literally read about the “666” in the book of Revelations and thought, “Man this is a good idea! We could control everyone that way. How do we get them to accept it? Scare them into thinking they are all going to die despite a general population mortality risk of only 0.009% over the course of Pfizer’s study”
Indeed that appears to be the only reason to rush this BLA. The drug is widely available, and the FDA is letting public health officials get away with deceptive marketing campaigns that would SLAPPED DOWN hard if a pharmaceutical or supplement company sold things that way. “X number of unvaccinated people died in X area this week. It did not have to be this way!” when Pfizer has not even generated the data to show it has an impact on deaths (deaths were actually higher by 1 in the vaccine arm as you can see above in Table S4).
It was the legal push-backs against employers mandating an experimental vaccine that clearly affected the timing of this approval. This did not meet the normal standards for a vaccine approval, not by a long shot, and everyone knows it. The FDA should have taken their time to do things correctly, and not make obvious errors that make is clear to everyone this was a political decision. And Pfizer should not have rushed to cross everyone over so as to prevent there being any control group to truly be able to objectively assess both long term safety and efficacy. All remaining followup data is not just as anecdotal and unscientific as the VAERS data that the scientific community loves to discount.
And this was not even the first time this happened with this vaccine. The vaccine never even met the minimal standards set forth in the October 22, 2020 meeting for an EUA. The FDA also allowed Pfizer to define a primary that was not a realistic measure of the use of the product. Unfortunately for them, that same choice on the severe COVID-19 secondary endpoint came back to bite them, or I should say should have come back to bite them if the FDA were being objective on the original EUA. The FDA panel in December never had a substantive discussion of the primary two risks of this vaccine (1) the pathogenic priming / immune enhancement that prevented a safe vaccine from ever even progressing into humans for the related SARS-CoV1 virus despite the better part of two decades of research and (2) the long term safety of a completely novel set of vaccines that have never been used in large numbers of healthy patients before. I plan on putting up future articles on each item mentioned in this paragraph because they deserve their own discussions.
The FDA spent most of time discussing how to continue the follow up given Pfizer told the panel they would be prospectively contacting patients telling them they had a right to get the vaccine when the EUA was approved and available for their age/risk group.
What the FDA panelists should have done at that point was was say, “There is a substantive public interest in knowing the exact and safety and efficacy profile of this vaccine. Based on the limited number severe COVID-19 cases in the placebo arm as per the pre-specified secondary endpoint (exactly 3 in the placebo arm, see Table 11 above) and there being no deaths in the trial data we have seen, there is no justification for an EUA because a randomized controlled trial is showing the estimates of COVID-19 morbidity presented to the FDA panel by government authorities are too high by almost an order of magnitude and the overall mortality risk from this disease is relatively minimal. This is not that pressing of a public health risk relative to the dangers from the mass use of therapeutic treatment with an unknown long term safety and efficacy profile. This is especially true given the fact Pfizer is admitting that if we grant the EUA, it will basically prevent us from having blinded long term follow up data to actually answer the questions of whether the risks outweigh the benefits.” As promised, the data now is completely unblinded due to massive crossover. Now, we will never have good randomized long term safety and efficacy data on this vaccine. Any safety or efficacy data will be difficult to characterize because there is no control group to assess the unvaccinated.
The most intelligent comment on the subject was by one reviewer who told Pfizer that it is in your best interest that the follow-up continue blinded because the number of cases prevented would increase relative to short term side effects (improving the risk reward calculus), you can show how durable the response is. Then people will feel less hesitant to get the vaccine with real randomized long term data, without getting the (correct) impression, this was rushed to market.
And with the publication of the 6 month data, we now know, based on the data we have (we never got the final severe adverse event data in the label), that the absolute difference in severe adverse events over baseline for the vaccine was more than 3 times the absolute difference in baseline for severe COVID-19 cases. And, at the end of the day, that is what you care about – the number of severe events prevented is more than the severe adverse reactions. But your odds of an adverse severe outcome are actually 41.6% worse if taking the vaccine based on the data in the 6 month preprint but hidden from the label! And, the difference seen in the trial highly statistically significant by my math.
It should be the burden of the manufacturer to prove the opposite, a statistically significant benefit in severe disease prevented over severe adverse events, and if the only sample shows a 41.6% increase in severe outcomes over placebo, an approval should not even be contemplated.
Some people would argue that severe adverse events are not the same as severe COVD-19. That is true. But how do you know which is more severe? A non-serious pulse oximeter reading of 92 with no ICU or hospital visit can be considered “severe” from the protocols set forth in the study. And that would be “severe,” not serious. Would you rather have that or 5 months and counting of your life with constant pain worse than you have ever had like Sally Kirkland right after her second jab? I would rather survive the ICU with critical COVID-19 than have a permanent vaccine injury like that. Some would probably prefer death over constant pain! An injury does not have to be “serious” or “life-threatening” to make life unbearable.
How would Pfizer label this? Given everything I have seen from Pfizer, if I were a betting man I would say “severe adverse event” not “serious” because it is not life threatening and it is in their best interest to most diminish any real life impairment that could potentially be considered “serious” under an FDA standard definition. After all, they apparently never even intended to disclose severe adverse events on the label since that information still is missing. It thus to their advantage to have a bias towards “severe” over “serious.” And we still do not know the final number on “severe adverse events,” only the data presented in the recent pre-print which was not complete based on updated data on total adverse events from the label.
This is why trust in the FDA is crucial. We are supposed to rely on our public health officials to hold these people accountable. Just based on the information we have on severe events without detail, objectively this drug should have never been approved – the important treatment effect was too low, allowing severe adverse events to outnumber severe COVID-19 cases and the absolute numbers of death to be higher in the vaccine arm. And that is before we even talk about long term follow up data and pathogenic priming fears given what we know about attempts at creating vaccines in SARS and MERS.
Given the fact that the FDA let Pfizer publish what appears to be blatantly false data on its secondary endpoint on the label, no one should have any confidence that any of this data has not been thoroughly massaged by Pfizer with no push-back from reviewers.
Until we have 100% transparency into the data (particularly all the adverse events), a truly independent assessment thereof, and the proper long term follow up necessary for a treatment given to healthy patients to prevent an observed COVID-19 death rate of 0.009% over 4.3 months, no one should trust this vaccine, only that it was intentionally rushed through the process with no objective analysis or adherence to normal scientific procedures.
This is not Ebola. 0.009% is not a death rate that scares me. What scares me is what I am not being told and what the true motives are for going through this fear mongering charade, not supported by the randomized controlled data we have.
When government authorities make Alex Jones look like a genius by spreading what is obvious disinformation and implementing the same heavy-handed policies he predicted they would do for years, it is no surprise that people have taken all sorts of crazy anti-vaccine stories (including some people saying viruses do not even exist at all!) and they become credible in the eyes of the scientific illiterate. The scientific community and an overly biased media with their big tech enablers have no one to blame but themselves for the fact up to half the population are distrusting much of what they say while they often spread scientific misinformation and uncollaborated conspiracy theories as if it has to be true.