
By Explicit Standards Set By the FDA on 10/22/2020 Pfizer's EUA Should Have Never Been Granted
The FDA had specific expectations on severe COVID-19 that were explicitly not met by Pfizer
The FDA set a very low bar to receive an Emergency Use Authorization (EUA). Their COVID-19 did not even meet these standards, and the FDA gave them the EUA anyway.
On October 22, 2020, an FDA advisory committee was commissioned to set out the minimum standards for granting an EUA. Here is the slide from the presentation that I want to focus on:

Source: LICENSURE AND EMERGENCY USE AUTHORIZATION OF VACCINES TO PREVENT COVID-19: CLINICAL CONSIDERATIONS, Slide 17
The third bullet sets out that the manufacturer should demonstrate “Sufficient cases of severe COVID-19 in placebo recipients, collected in the same timeframe as primary endpoint cases, to assess case split between vaccine vs. placebo groups as signals for the effectiveness and for ERD”
“Collected in the same timeframe as primary endpoint cases” essentially means you are using the same time criteria as you used for assessing your primary endpoint to collect these cases. In this case, as discussed elsewhere on this site, that was 7 days after the second dose. The primary analysis was set to maximize the efficacy of the drug by abandoning “intent to treat” from the first dose and focus only on those people who had not come down with COVID-19 before 7 days after dose 2. On top of that, they also excluded people who showed evidence of prior infection to further boost their chance of a positive result.
So to match this time frame and analysis a hard secondary endpoint was set for severe COVID-19 that matched the same patient population as the primary endpoint. This was in large part was to demonstrate what was required for the third bullet point on severe COVID-19 cases.
Here is the data as it was presented to the FDA panel as a briefing document for the December 10, 2020 meeting:
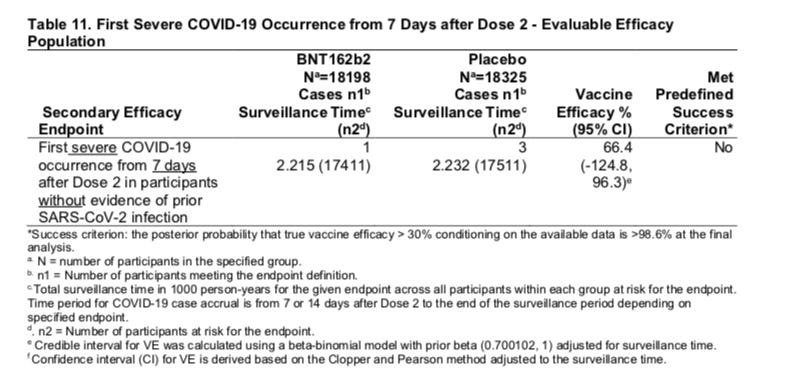
Source: VRBPAC FDA Briefing Document Pfizer-BioNTech COVID-19 Vaccine, page 31, Table 11
You can see this is highlighted as “Secondary Efficacy Endpoint” denoting this was the hard endpoint negotiated with the FDA and that it involved looking at “First severe COVID-19 occurrence from 7 days after Dose 2 in participants without evidence of prior SARS-CoV-2 infection” just like the primary endpoint.
The number of placebo patients with severe COVID-19 was a whopping three. Now I think that anyone who knows math knows three cases is not a representative enough sample to show practically anything. There is no way they met this criteria.
But just to prove the point, the FDA did provide some additional guidance as to what they thought would be sufficient cases at the 10/22/2020 panel meeting. It was woefully inadequate, but this vaccine trial missed it.
Thirdly, we expect sufficient cases of severe COVID-19 in placebo recipients, cases that have been collected in the same timeframe as primary endpoint cases, so that we can assess the case splits between vaccine and placebo groups looking for signals of both vaccine effectiveness against severe disease and also for enhanced respiratory disease.
In our guidance document, we mentioned five cases in the placebo group as being generally sufficient to meet this expectation. However, in cases where the vaccine efficacy point estimate and lower bound are both exceptionally high and there are no severe cases in the vaccine group, fewer than five cases may be acceptable.
Source: 10/22/2020 FDA Panel Meeting Transcript, pages 191-192
So you can see five cases is all they required in a study with over 20,000 patients enrolled in each arm, and they still did not have enough cases to reach that low-ball hurdle. Now they did say the number of cases may be below five if the efficacy was high and there were no severe cases in the vaccine arm. But there was one severe case in the vaccine arm.
In the most explict terms possible the sponsor completely missed the requirements for an EUA!!
At the panel meeting on December 10th they were even asked if they had additional data to present because it was obvious they missed the prespecified secondary endpoint. They had no additional data to present other than reporting on severe COVID-19 cases in the placebo group outside the window they had agreed upon.
In fact even when they expanded the group to include those with prior COVID-19 exposure, they only came up with four instead of five in the prespecified window terminating at 7 days after dose 2:

Source: VRBPAC FDA Briefing Document Pfizer-BioNTech COVID-19 Vaccine, page 31, Table 11
The bottom line is that they did not meet the prespecified criteria and the FDA panel overwhelmingly voted to recommend granting the EUA anyway!
The fix was in for a multibillion dollar company like Pfizer.
Why this Matters
Most obviously, it shows that the FDA is not an independent arbiter of consumer safety. They did not take their own standards seriously. Indeed, to anyone who listened to that December 10, 2020 panel meeting, it was very obvious it was a rubber stamp with virtually no attention paid to the most important risks such as pathogenic priming, the reason no coronavirus vaccine has ever been successfully produced in the past. The panel literally spent most of the day talking about how to deal with the fact Pfizer would be reaching out to the people in the trial telling them they can get the vaccine as soon as the EUA was approved and how to handle that particular situation.
But there is something else that is important about why you would require a certain number of severe events in the placebo arm. The most obvious is to show efficacy of the treatment against severe COVID-19 to a reasonable degree of confidence. But additionally the EUA is only supposed to be granted for pressing needs. It literally is supposed to be an “emergency” use authorization.
3 cases out of 2,232 patient years shows the one year risk of severe disease to be only 0.13%. Does this sound like an emergency? By contrast the estimates presented by regulatory officials to the FDA panel on December 10, 2020 suggested the risk per 6 months was supposed to be 1 in 200, or 1% annual risk. That is an order of magnitude over what was observed here. Even if you take the entire cohort presented to the panel back to dose 1, you are only looking at a 9/4006 = 0.25% risk of severe COVID-19 disease, still four fold less than the estimates. And there were no COVID-19 deaths at all in either arm.
The trial clearly showed that COVID-19 was not nearly as bad as estimated. Remember the declaration that the EUA was appropriate was made in the spring of 2020 when doctors were literally killing people because they put them prematurely on ventilators and had no idea how to treat the disease. A lot of people were panicking because they saw videos coming out of Wuhan China showing people just falling over dead in the streets from COVID-19. Ever see this anywhere else?
Even if you take the latest Pfizer vaccine data which the panel did not have then, you have an annualized risk that is 30/8,288 = 0.36% risk of severe COVID-19 at best, and a 0.025% annualized risk of death from COVID-19.
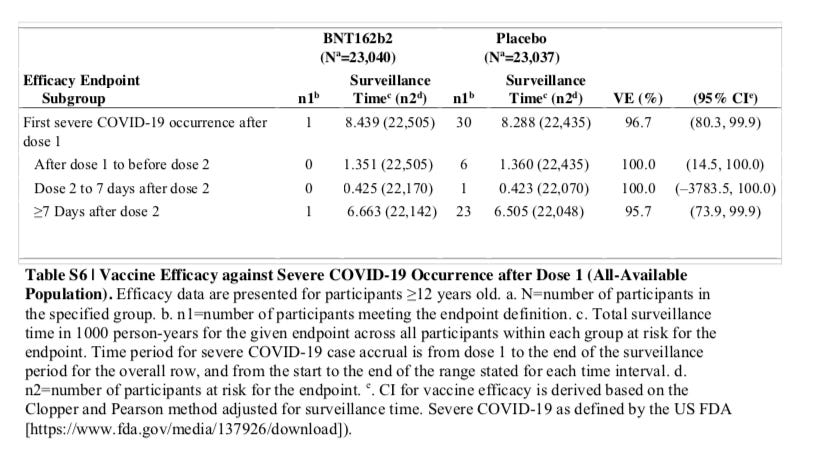
Source: SUPPLEMENTARY APPENDIX to Six Month Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine, Page 15, Table S6
Neither of these is anywhere near the projections and neither justifies an EUA. The long term side effects of any therapeutic treatment could easily be greater than that, and we know from the last data we have, the severe adverse events are much higher than this at 1.2%, and the excess deaths of vaccine over control in the under 5 months of followup is at 0.18%, double the rate of COVID-19 deaths in the placebo arm during the same period of time.
If this was the lowly standard for an EUA, then we should have an EUA for virtually every drug in existence! Especially stuff like heart disease and cancer which are way more lethal. The EUA was meant for major pandemics and bio-weapons with a very high mortality, not some bug that kills less than 0.1% of the entire population on an annual basis. Why have an FDA at all if anything with a 0.025% death rate in the target population is an emergency that allows you to forgo all typical testing? Just rush everything out in less than a year!
No rational person who looks at the data over the media and government fear porn would take this seriously as a risk. Heart disease, cancer, etc. are much bigger concerns.
And that is why the FDA number of five was even too low. Maybe the panel could he issued a conditional approval on meeting the five patient standard in a few weeks. That would have been at least intellectually consistent. But even then, the FDA standard was too low and did not recognize that an absurdly low number of cases meant (1) their estimates were too high to begin with and (2) there was thus no “emergency” to begin with.
We Might Never Know the Real Safety and Efficacy Because the FDA Rubber Stamped this EUA
The December 10th panel was well aware of the difficulties of continuing to gather long term follow up data. Pfizer basically told the panel they had some sort of moral obligation to tell people in trial the EUA was approved and that they could the vaccine if they wanted to. And most of what they discussed was how to manage this so they can get the best data possible even though the trial had been thoroughly crossed over. This shows they knew there was going to be a problem in granting the EUA in terms of maintaining the integrity of the trial.
The problem is, there was no moral obligation. The trial showed, if anything, the COVID-19 risk was over-hyped by the media and governments around the world. These were brand new vaccine types (mRNA, viral vector) that had never been used in large numbers of otherwise healthy individuals before. If using a vaccine in a cancer patient who is on chemo-therapeutics and having a compromised immune system, not only might the vaccine have different effects in the body, but lower level side effects could be masked by a very high background in patient morbidity. As one panel mentioned, even the 1 in 200 and 1 in 1000 risk numbers were not exactly high in terms of creating some moral obligation.
Society has a much more pressing need in getting clinical data on the long term safety and efficacy of these products. We only now have randomized efficacy data for about 4.5 months in the enrolled patient population. Some subgroups will be greater than that, but probably not much more because after the EUA was approved, placebo patients started to get the vaccine in mass. Without a placebo arm, you are never going to have a background arm to show excess numbers of something like cancers or cardiac morbidity over control as time goes on. You are not going to know what the infection rate of the vaccinated versus the unvaccinated is over time, which might be important in assessing real world antibody dependent enhancement as new variations arise.
The EUA literally is going to prevent us from ever identifying in a rigorous manner the long term problems if they are subtle. And subtle matters when you are talking giving a medicine to healthy persons, especially children who are even a smaller risk for COVID-19 complications. A myocarditis risk of only 200 per 1 million in 16-17 year olds it too much to justify giving a vaccine against a disease they have an even much lower risk of dying from!
What the panel members should have said is, “Well Pfizer, since you are so adamant about thinking the existence of the EUA mortally obligates you to inform people they can get the vaccine, then I have a moral obligation not to approve this EUA to make sure we get a full two year’s of data to make sure this is safe and effective.” I bet Pfizer would have quickly changed its tune then. The FDA should have also made it clear, if no blinded two years of follow up was presented because of massive crossover, no BLA would be issued due to lack of data. But the FDA is so captive to industry, that was never going to happen. A weak and obsequious FDA lets the pharmaceutical manufacturers get basically what they want.
But the EUA for all of these vaccines should have been rejected out of hand due to these trials showing COVID-19 is a risk that is well below the potential long term side effect risk of any therapeutic. This was not Ebola! This was not such an “emergency” that we throw out the window all principles of evidence based medicine such as animal trials, adequate phase 2 trials, and long term follow up.